1. DNA Replication and epigenetic inheritance
Our lab investigates how cells preserve epigenetic information during DNA replication, especially under replication stress. While genome duplication copies DNA sequence, maintaining cell identity also requires faithful transmission of chromatin states—particularly histone post-translational modifications (PTMs)—from parental to daughter strands. Disruption of this process contributes to epigenetic erosion in aging and fuels cancer initiation, heterogeneity, immune evasion, and therapy resistance.
A key mechanism of epigenetic inheritance is parental histone recycling: as replication forks progress, parental nucleosomes are disassembled ahead of the fork and then redeposited onto nascent DNA, where inherited marks help re-establish the local PTM landscape after replication. Replication stress complicates this choreography. Under stress, forks can undergo fork reversal and cells can accumulate single-stranded DNA (ssDNA) gaps—events that are common but whose consequences for epigenetic inheritance have been poorly defined.
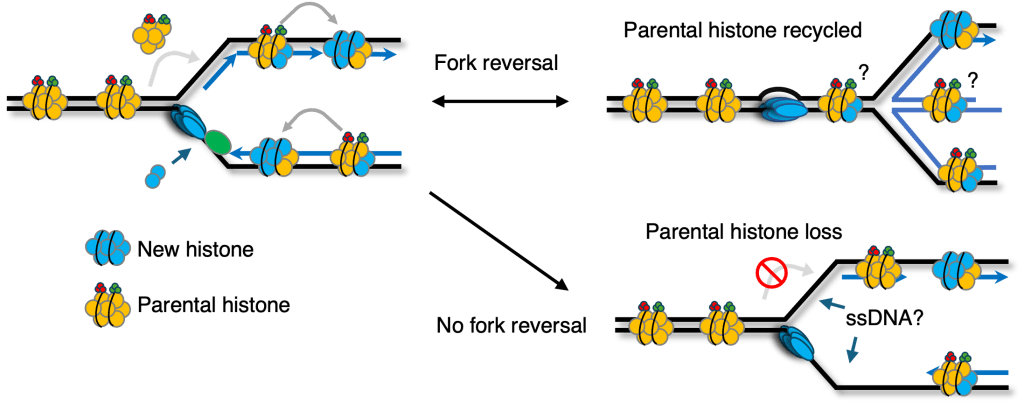
Our preliminary work addresses this gap by proposing and supporting a new concept: fork reversal helps safeguard epigenetic stability during DNA replication under stress. In fork-reversal–deficient cells, we observe reduced nucleosome density on nascent DNA together with loss of parental histone PTMs (including canonical heterochromatin-associated marks). These findings provide a mechanistic bridge between replication-fork remodeling and epigenetic maintenance, and are consistent with the broader idea that replication stress should be viewed not only as a DNA-centric threat, but also as a driver of epigenetic instability with disease relevance.
To connect mechanism to biology in a quantitative and fork-proximal manner, our lab combines complementary replication-fork multi-omics and chromatin assays. We use iPOND-SILAC-MS to measure proteins bound to nascent DNA and reveal chromatin assembly defects in fork-reversal–deficient settings. We validate nucleosome loss genome-wide using ChOR-seq, a sequencing-based method that profiles histone occupancy on newly synthesized DNA; these data confirm diminished histone H3 signal on nascent DNA and support a model in which excess ssDNA—normally suppressed by fork reversal—compromises nucleosome density.
We also develop approaches to directly read out histone PTMs at replication forks. By coupling iPOND-MS with histone propionylation, we enable sensitive, high-throughput detection of fork-proximal histone modification patterns. Using this strategy, we find a consistent shift: parental-histone marks decrease while new-histone marks increase in fork-reversal–deficient cells, supporting impaired parental histone recycling during stressed replication.
Overall, our research defines a framework linking fork dynamics (fork reversal and ssDNA gaps) to faithful epigenetic transmission, with direct implications for understanding how replication stress promotes epigenetic drift and how tumors with fork-reversal defects may exhibit distinctive vulnerabilities and treatment responses.
2. Replication fork proteomics to identify new therapeutic targets
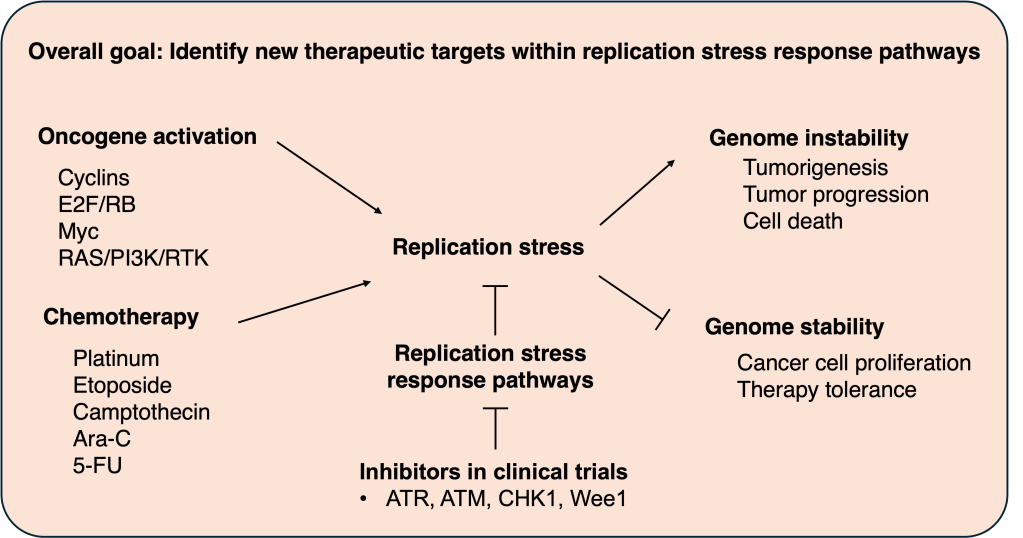
Our studies focus on how cells cope with replication stress—a pervasive challenge in both normal proliferating tissues and cancer. Replication stress can be triggered by oncogene-driven hyperproliferation and by frontline chemotherapies that directly perturb DNA synthesis, leading to genome instability that fuels tumorigenesis, tumor progression, and treatment outcomes. Because cancer cells rely heavily on replication stress response pathways (e.g., ATR–CHK1 signaling) to survive, our overarching goal is to identify new, actionable therapeutic targets within these pathways and to inform rational combination strategies beyond current DDR inhibitors.
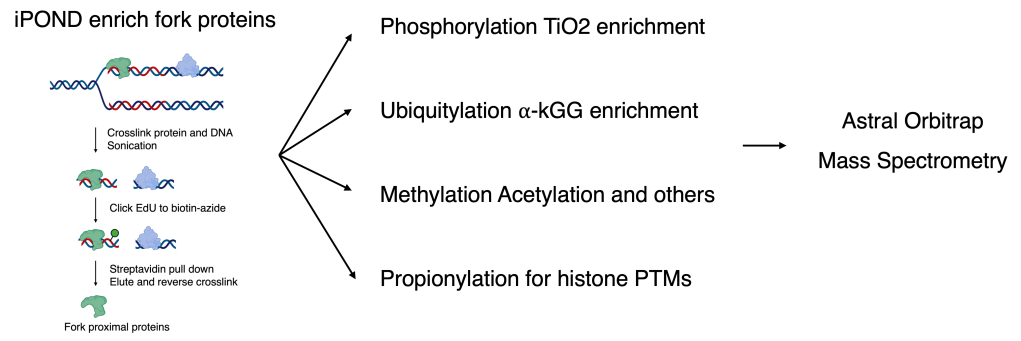
iPOND: Isolation of Proteins On Nascent DNA
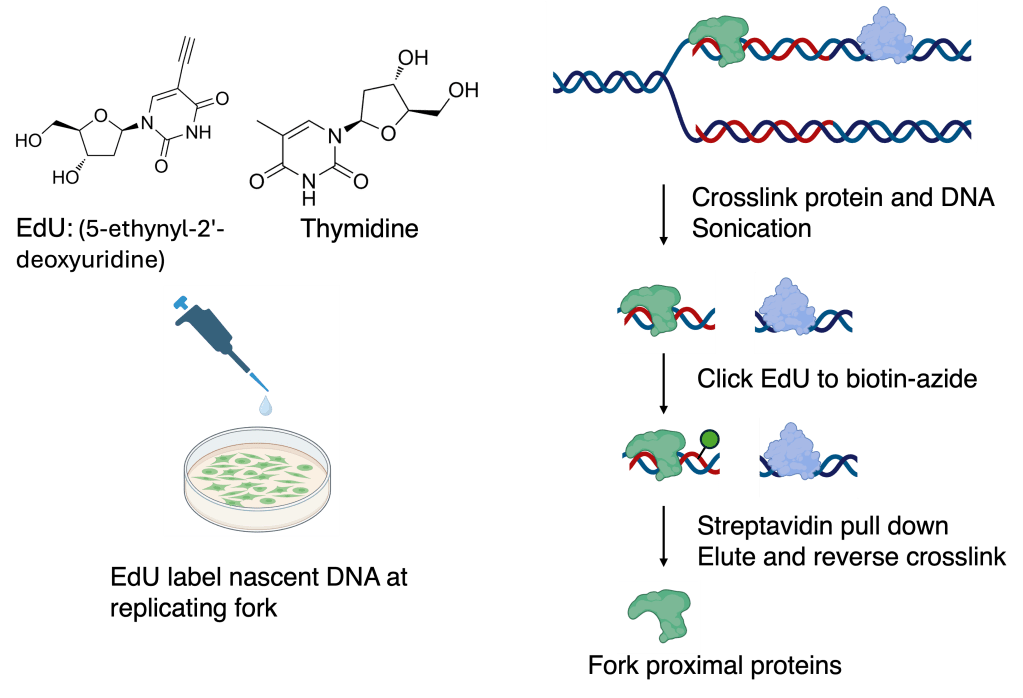
Stress responses at stalled replication forks are governed not only by the recruitment of repair and signaling factors, but also by dynamic protein post-translational modifications (PTMs)—including phosphorylation and ubiquitylation—that tune protein activity, interactions, and turnover. To capture this regulation in its native fork context, we optimized an iPOND-based fork proteomics workflow coupled to tandem PTM enrichment: nascent DNA is labeled with EdU, fork-proximal proteins are isolated via click chemistry, and quantitative mass spectrometry is used to measure stress-induced changes. Building on this platform, we generate a fork-proximal PTM atlas by enriching phosphopeptides (TiO₂), ubiquitinated peptides (diGly capture), and profiling histone PTMs at high resolution using histone propionylation chemistry. Together, these approaches have already uncovered tens of thousands of fork-associated PTM events, creating a powerful resource to identify new regulatory nodes and therapeutic opportunities in replication stress biology.